Die Untersuchung von Abortmaterial beginnt mit der Teilung der Probe. Aus einem Teil wird DNA isoliert, während der andere Teil kultiviert und für die Chromosomenanalyse verwendet wird. Wenn diese Analyse einen unauffälligen weiblichen Karyotyp ergibt, schließt sich eine PCR-basierte Mikrosatellitenanalyse an. Dabei wird das Abortmaterial mit dem EDTA-Blut der Mutter verglichen, um eine Kontamination des Untersuchungsmaterials mit mütterlichem Gewebe auszuschließen. Gelegentlich ist die Kultivierung des Abortmaterials nicht erfolgreich. Für eine erfolgreiche Untersuchung sind mindestens 10 mg Abortmaterial in einem sterilen Probengefäß mit 0,9 % NaCl-Lösung erforderlich.
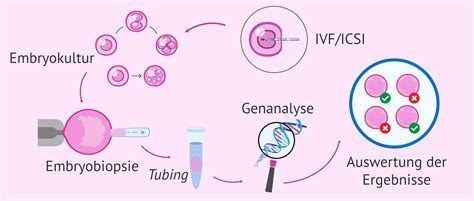
Methoden der genetischen Diagnostik
Array-CGH (Array-based Comparative Genomic Hybridization)
Die Array-CGH ist eine Analysemethode, bei der eine simultane Hybridisierung von fluoreszenzmarkierter Patienten- und Kontroll-DNA auf einem Trägerchip mit DNA-Fragmenten erfolgt. Mittels dieser Methode lassen sich Kopienzahlveränderungen der DNA nachweisen, die aufgrund ihrer Größe in der konventionellen Chromosomenanalyse nicht erkannt werden können. Es kann genomweit ein Verlust von genomischen Bereichen (Deletion) und Hinzugewinn (Duplikation) detektiert werden. Die Veränderungen können in ihrer Größe sehr variabel sein und einige hundert bis mehrere Millionen Basenpaare umfassen. Methodisch bedingt können balancierte Chromosomenveränderungen (z.B. Inversionen) nicht erfasst werden. Häufige Anforderungsgrunde für die Array-CGH-Diagnostik sind Patienten mit mentaler Retardierung und/oder Organfehlbildungen und Dysmorphiezeichen. Indikationskriterien sind eine „Geistige Entwicklungsstörung ungeklärter Ätiologie“ oder autistische Verhaltensweisen.
Fluoreszenz in situ Hybridisierung (FISH)
Die Fluoreszenz in situ Hybridisierung (FISH) ist eine Methode zum Nachweis von Nukleinsäuren (in der Regel DNA) in einzelnen Zellen oder auf Chromosomen. Dabei wird eine künstlich hergestellte Sonde aus Nukleinsäure eingesetzt, die über Basenpaarungen an die nachzuweisende Nukleinsäure bindet (Hybridisierung). Der Nachweis wird direkt in der jeweiligen Struktur (in situ) durchgeführt. Für die FISH-Untersuchung werden etwa 10 mg Chorionzotten in einem sterilen Probengefäß (0,9 % NaCl-Lsg.) benötigt.
SSP-PCR und Fragmentanalyse
Die SSP-PCR (Sequence-Specific Priming Polymerase Chain Reaction) und die Fragmentanalyse sind weitere molekulargenetische Methoden, die zur Detektion spezifischer Genmutationen eingesetzt werden. Sie finden Anwendung unter anderem bei der Untersuchung von Männern mit Fruchtbarkeitsstörungen, insbesondere bei der Abklärung von Mikrodeletionen auf dem Y-Chromosom (AZFa, AZFb, AZFc) oder bei der Analyse des CFTR-Gens im Rahmen der Cystischen Fibrose.
Genetische Syndrome und Erkrankungen
Amelogenesis imperfecta
Amelogenesis imperfecta ist eine erblich bedingte Störung der Zahnschmelzbildung. An diesem Prozess ist eine Vielzahl von Proteinen beteiligt, die bei Mutationen zum klinischen Bild der Amelogenesis imperfecta führen. Abhängig davon, welches Protein durch eine Veränderung in seiner Funktion beeinträchtigt ist, kommt es zur Ausprägung von verschiedenen klinischen Symptomen.
Arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC)
Die arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC) ist durch einen fortschreitenden fibrillären Ersatz des Myokards gekennzeichnet, der bei jungen Menschen und Sportlern zu ventrikulären Tachykardien und plötzlichem Tod führen kann. Sie betrifft in erster Linie den rechten Ventrikel, kann aber auch den linken Ventrikel mit einbeziehen. Das Krankheitsbild ist selbst innerhalb von Familien sehr variabel und folgt typischerweise einem autosomal dominanten Erbgang mit unvollständiger Penetranz. Es wird angenommen, dass 30-80% der Erkrankungen auf eine genetische Prädisposition zurückzuführen sind. Die häufigsten genetischen Ursachen sind Mutationen in den Genen DSC2, DSG2, DSP, JUP, PKP2 und TMEM43. In ca. 30-50% der ARVC-Patienten können Mutationen in desmosomalen Genen festgestellt werden.
Chorea Huntington (Morbus Huntington)
Die Chorea Huntington ist eine Erkrankung des Nervensystems mit einer geschätzten Prävalenz in Europa von etwa 5 auf 100.000 Einwohnern. Erste Anzeichen treten meist zwischen dem 35. und 45. Lebensjahr auf. Als Frühsymptome können psychische Veränderungen auftreten. Im Weiteren ist die Huntington-Krankheit durch unwillkürliche choreatische Bewegungen gekennzeichnet und führt zu einem vollständigen Verlust von motorischer Kontrolle und intellektuellen Fähigkeiten (Demenz). Das verantwortliche Gen für die Chorea Huntington ist auf Chromosom 4 lokalisiert. Im sogenannten Huntingtin-Gen liegt bei den Anlageträgern bzw. den Betroffenen eine Verlängerung einer kleinen Einheit des Erbmaterials, der Basenabfolge CAG, vor. Die Erkrankung wird autosomal dominant vererbt.
Cowden-Syndrom (CS)
Das Cowden-Syndrom (CS) ist Teil des ‚PTEN Hamartom Tumorsyndroms‘ (PHTS) und wird autosomal dominant vererbt. Das mit CS verbundene erhöhte Risiko umfasst Brust-, Endometrium-, Schilddrüsen-, Dickdarm- und Nierenkrebs. Keimbahnmutationen im Tumorsupressorgen PTEN sind ursächlich für das CS. Bestimmte dermatologische Befunde (einschließlich multiple Trichilemmome, orale Papillome und akrale Keratose) können auf das Vorliegen eines CS hinweisen. Auch eine makuläre Pigmentierung der Glans penis ist ein wichtiges Diagnosekriterium bei männlichen Patienten. CS-ähnliche Symptome können mit Mutationen in anderen Genen oder einer Promotor-Methylierung des KLLN-Gens vergesellschaftet sein.
Cystische Fibrose (Mukoviszidose)
Die Cystische Fibrose ist eine der häufigsten Stoffwechselerkrankungen in Populationen kaukasischer Abstammung und wird autosomal-rezessiv vererbt. In Deutschland tritt sie mit einer Häufigkeit von etwa 1:3300 Neugeborenen auf. Bei diesem Krankheitsbild handelt es sich um eine generalisierte Dysfunktion der exokrinen Drüsen, die im klassischen Fall Atemwege, Verdauungssystem und Reproduktionstrakt betreffen kann. Der Erkrankung liegt eine Funktionsstörung eines Chlorid-Ionenkanals zugrunde. Der Übergang zwischen typischer CF, milden und aberranten Formen sowie asymptomatischen Verläufen ist fließend. Eine genetische Erkrankung, die bei Männern zu Unfruchtbarkeit führt, ist die Mukovizidose. Auch durch heterozygote Mutationen im CFTR-Gen kann es zu ein- oder beidseitiger Aplasie der Samenleiter kommen (CUAVD bzw. CBAVD).
DiGeorge-Syndrom
Das DiGeorge-Syndrom beruht auf einer gestörten Entwicklung der dritten und vierten Kiemenbogentasche, was zu einer Anlage-/Entwicklungsstörung einer Reihe von Organen führt. Das klinische Spektrum dieser Störungen ist außerordentlich breit. Der Begriff DiGeorge-Syndrom wird für Kinder verwendet, die sich in der Neonatalperiode mit typischem Herzfehler, Thymushypoplasie und Hypocalzämie präsentieren. Das Velocardiofaciales (Shprintzen)-Syndrom wird für ältere Kinder verwendet, bei denen die nasale Sprache durch velopharyngeale Inkompetenz im Vordergrund steht. CATCH22 ist ein kollektives Akronym für die unterschiedlich ausgeprägten Syndrome. Das Spektrum des Immundefekts durch die Thymushypo- oder aplasie ist ebenfalls breit. Die meisten Formen des DiGeorge-Syndroms sind mit einer Mikrodeletion der Region 22q11.2 verbunden.
Dilatative Kardiomyopathie (DCM)
Die Diagnose einer Dilatativen Kardiomyopathie (DCM) wird gestellt, wenn eine Vergrößerung des linken Ventrikels sowie eine systolische Dysfunktion vorliegt. Eine arrhythmogene rechtsventrikuläre Kardiomyopathie (ARVC) mit vorherrschender linksventrikulärer Beteiligung kann sich möglicherweise als DCM präsentieren. Die DCM manifestiert sich in der Regel erstmals bei Erwachsenen im 4. bis 6. Lebensjahrzehnt. In der Mehrzahl der Fälle werden pathogene Mutationen in verschiedenen Genen detektiert (z.B. TTN, LMNA, MYH6/MYH7). Mutationen in mehr als 30 Genen wurden bei bis zu 30-35 % der Personen mit familiärer DCM identifiziert.
Katzenschrei-Syndrom (Cri-du-chat-Syndrom)
Das Katzenschrei-Syndrom oder Cri-du-chat-Syndrom ist erstmals 1963 beschrieben worden und benannt nach dem katzenähnlichen Schreien der betroffenen Kinder im frühen Kindesalter. Die Ursache des CDC-Syndroms ist eine partielle Deletion am kurzen Arm eines Chromosoms 5 (partielle Monosomie). Der Verlust erfolgt in der Regel zufällig. In 15 % der Fälle wird das CDC-Syndrom durch eine unbalancierte Chromosomentranslokation ausgelöst, wobei bei 10 % der Kinder bei einem Elternteil bereits ein Teil des entsprechenden Chromosomenarmes abgebrochen ist und sich an einem anderen Chromosom angelagert hat.
Fruchtwasseruntersuchung und Chorionzottenbiopsie
Für die Untersuchung von Fruchtwasser werden etwa 10 mg Chorionzotten in einem sterilen Probengefäß (0,9 % NaCl-Lsg.) benötigt. Die Vorläufige Befundung aus der Direktpräparation erfolgt ca. 1-2 Tage, der Endbefund ca. 14-18 Tage. Bei Fruchtwasserproben erfolgt ein vorläufiger Befund ca. 6-9 Tage und ein Endbefund ca. 14-18 Tage.
Unfruchtbarkeit beim Mann (Oligo-/Azoospermie)
Etwa 50% aller infertilen Männer weisen eine deutliche Reduktion ihrer Samenzellen auf (Oligo- oder Azoospermie), wobei die kausalen Ursachen vielfältig sind. Es wird zwischen obstruktiver und nicht-obstruktiver Azoospermie unterschieden. Bei 10-20% der Patienten mit nicht-obstruktiver Oligo- oder Azoospermie ist die Ursache der Infertilität durch das Auftreten von drei verschiedenen Mikrodeletionen auf dem langen Arm des Y-Chromosoms assoziiert (AZFa, AZFb und AZFc). Männer mit einer kompletten AZFa-Deletion weisen keine Keimzellen mehr in ihrem Hodenepithel auf. Liegt dagegen eine partielle AZFa, AZFb, oder eine komplette AZFc-Deletion vor, können noch reife und befruchtungsfähige Spermien im Hodenepithel vorgefunden werden. Es ist deshalb für den Patienten wichtig zu wissen, ob die Ursache der Fertilitätsstörung eine komplette AZFa oder komplette AZFb/c-Deletion ist.
Male Infertility: Your Guide to Y Chromosome Microdeletions
Hautfibroblasten / OP-Material
Für die Untersuchung von Hautfibroblasten oder OP-Material werden mindestens 15 mg in einem sterilen Probengefäß (0,9 % NaCl-Lsg.) benötigt.